【7】電子密度,静電ポテンシャル,波動関数の三次元可視化
- 〜いよいよ計算結果の電子密度,静電ポテンシャル,波動関数を三次元可視化します。〜
- ◎1.以下は、自分の計算環境がc:\dvxa\calc\○○、ノンスピン版でクラスgという例で説明しております。
- ◎2.c:\dvxa\calc\○○では、DV-Xα計算が収束して、
f01 ・・・アスキーファイル、原子の座標等。
f09 ・・・バイナリファイル、固有ベクトルの値。
f39 ・・・バイナリファイル、全変数配列の値。
がすでにある状態です。この3つのファイルは、以下の作業に必ず必要です。
- ◎3.contrd.tbzを解凍後のフォルダに入っているcontrd\c04d を 自分の計算ディレクトリ (c:\dvxa\calc\○○)にコピーしてください。
- ◎4.F08Eを見て、図を描きたい分子軌道(波動関数)の番号をメモしておいてください。波動関数の番号については、黄色い本「はじめての電子状態計算」の第7章-5 「波動関数・電子密度などの等高線図の作図をする」を参照してください。プログラム【WAVNUM】を利用すると便利です。
- ◎5.例えば、135, 136, 137, 138 番目の分子軌道を描く場合は、c04d は、以下のようにすればOKです。

もし分子が巨大で、20a0×20a0×20a0に入りきらない場合は、例えば、

というように、描画範囲を拡張してやってください。ここで指定する直方体(3辺が同じ長さの場合は立方体となります)の描画範囲は、DV-Xαで計算したモデルの全原子が完全に(余裕を持って)入り込んでいる必要があります。c04dで指定する描画範囲の数値、F05の座標数値は共に原子単位です。molda2dvで作成したF01の座標数値はÅ単位です。原子単位については、【6】DV-Xα分子軌道計算の【補足】を参照してください。
【補足】:F01に書かれている全原子の、(X座標の値、Y座標の値、Z座標の値)のそれぞれ最大値(プラス側の最大値)、最小値(マイナス側の最大値)をメモし、F01がmolda2dvで作られたものである場合は座標数値はÅ単位ですので、すべてのメモした値を0.529177で割って、原子単位に変換してみてください。
c04dの2行目で、原子単位の数値で指定する大きさの直方体は、その中に完全に(少しの余裕を持って)、F01のすべての原子の座標が入り込んでいる必要があります。
例えば、c04dの3行目が 0.0, 0.0, 0.0、2行目が 30.0, 25.0, 20.0 であった場合は、メモした原子単位でのX座標、Y座標、Z座標の最大値、最小値が、
X座標: (+15.0a0 〜 -15.0a0 )
Y座標: (+12.5a0 〜 -12.5a0 )
Z座標: (+10.0a0 〜 -10.0a0 )
の範囲に余裕を持って入り込んでいることをご確認ください。
これは、以下のように考えても構いません。
すなわち、原子単位の数値をÅ単位に直し(原子単位の数値に0.529177を掛けて)、
X座標: (+7.94 Å 〜 -7.94 Å )
Y座標: (+6.61 Å 〜 -6.61 Å )
Z座標: (+5.29 Å 〜 -5.29 Å )
の範囲にF01の全原子の全座標(Å単位)が余裕を持って入り込んでいることを確認すればよいことになります。
万が一この直方体から原子がはみ出していると、この後の◎8.において、VICSでF01を読み込んだときに、思いもかけないようなおかしな図になってしまいます(VICSでは、単位格子から原子がはみ出した場合、結晶学的な並進の操作が自動的に行われるため)。VICSはF01を読み込むときに、F01が置いてあるディレクトリに存在するc04dも、あわせて読み込みます。c04dで直方体の大きさを指定するときは、DV-Xα計算した全原子が完全に(少しの余裕を持って)入り込むようにご注意ください。
また、1行目は、どれだけ細かいメッシュで計算するかというメッシュの数なのですが、上の例では
100×100×100ですが、もっと細かく計算したい場合は、

ぐらいが、ほぼ最高に細かく計算させる場合の入力です。
これだと、かなりの計算時間がかかるうえ、生成されるファイルのサイズも何百MBという巨大なものになりますので、通常は51 51 51ぐらいか、あるいは101 101 101ぐらいが適当かと思います。
ただし【8】等電子密度表面上の静電ポテンシャルの三次元可視化を行うときには、251 251 251でCHG3D.sca, POT3D.scaを作成しておくと、とても美しい等電子密度表面を静電ポテンシャルの大小によって彩色した図を描くことができます。このことは、【8】等電子密度表面上の静電ポテンシャルの三次元可視化の【補足】に詳しく書いてあります。
【補足】:なお、このc04dを自動的に作成してくれるプログラムを大阪大学の水野正隆先生がご開発されています(c04dの2行目のx, y, z方向の長さ(直方体の大きさ)も、3行目の中心座標位置も、自動決定してくれます)。現在、DV-Xα研究協会の会員限定で公開されております。
- ◎6.c04dの準備ができましたら、
contrd g
というように、contrdをgクラスで回してください。なお、ここではgクラスで回していますが、分子の大きさやスピン分極を考慮するかしないかによって、このジョブクラスは変わってきます。ジョブクラスの選択については、黄色い教科書「はじめての電子状態計算」の付録B−5「変数のサイズについて」を参照してください。
かなり時間がかかると思いますが、contrdが無事終了したならば、
CHG3D.sca ・・・電子密度
POT3D.sca ・・・静電ポテンシャル
W135-3D.sca ・・・135番目の分子軌道の波動関数
W136-3D.sca ・・・136番目の分子軌道の波動関数
W137-3D.sca ・・・137番目の分子軌道の波動関数
W138-3D.sca ・・・138番目の分子軌道の波動関数
というファイルができているはずです。以上でcontrdの作業は終了です。
- ◎7.次は、まずVENUS.tbzを解凍した後に生成するVENUS\Programs\VICS.exeをダブルクリックしてVICSを立ち上げます(うまくいかないときは、VICS_P3.exeを起動してみてください)。
VICS: Menu
Dialog
Graphics
VICS
の4つのウィンドウが立ち上がります。
- ◎8.VICS: Menu の 【Open】をクリックし、
ファイルの種類(T):のプルダウンメニューで、下から2行目の【SCAT f01】を選択、
ファイルの場所(I):のプルダウンメニューで、c:\dvxa\calc\○○を指定し、
【f01】を選んで【開く(O)】をクリックします。
- ◎9.VICS: Menu の 【Bonds】をクリックし、
Add new bondsの欄で、どの原子とどの原子を結合の線で結ぶのかを指定します。
結合の指定は、以下に示す3種類の方法を繰り返して、あるいは組み合わせて(そのたびに【Add】をクリックすることにより)指定しますが、すべての指定が終わるまでは【OK】ボタンをクリックしてはいけません。描こうと思っている結合の指定をすべて【Add】し終わってから、最後に【OK】をクリックしてください。
結合の指定の仕方は、以下の3種類です。
(1)例えばA1原子からの距離1.4 Å以上、1.6 Å以下の結合を探して表示する場合は、
Search atoms bonded to A1
を選んで、A1:に該当する原子をプルダウンメニューで指定し、
Min. distance: 1.4
Max. distance: 1.6
で【Add】をクリックします。
(2)A1原子からA2原子への距離1.4 Å以上、1.6 Å以下の結合を表示する場合は、
Search A2 bonded to A1
を選んで、A1:とA2:に該当する原子をプルダウンメニューでそれぞれ指定し、
Min. distance: 1.4
Max. distance: 1.6
と距離の最小値、最大値を指定し、【Add】をクリックします。
(3)原子を特定せずに全体の中で距離1.4 Å以上、1.6 Å以下の結合を探して表示する場合は、
Search molecules
を選んで、
Min. distance: 1.4
Max. distance: 1.6
と距離の最小値、最大値を指定し、【Add】をクリックします。
すべての【Add】が終わってから、【OK】をクリックしてGraphics 画面で結合が生成したことを確認します。
もしおかしな結合が生えてしまった場合は、VICS: Menu の 【Bonds】をクリックし、
Search listの中からおかしな指定を選んで【Delete】をクリックすれば、おかしな結合を削除できます。
なお、随時、Graphics画面のマウス右側ドラッグで分子は好きなように回転できます。
- ◎10.最後に、VICS: Menu の【Save】をクリックし、
保存する場所(I): にc:\dvxa\calc\○○を選んで、
適当な名前を付けて、【保存(S)】をクリックして、
標準のVICSフォーマットで保存します(例えば、complex.vcs)。
VICS: Menu の【Exit】をクリックして、VICSを終了します。
- ◎11.次に、VENUS\Programs\VEND.exeをダブルクリックして、VENDを起動します(うまくいかないときは、VEND_P3.exeを起動してみてください)。
VEND: Menu
Dialog
Graphics
VEND
の4つのウィンドウが立ち上がります。
- ◎12.VEND: Menu の 【Open】をクリックし、
Format: のプルダウンメニューで、
上から5行目の【SCAT (*.scat, *.sca)】を選択、
【Browse...】をクリックし、
ファイルの場所(I): にc:\dvxa\calc\○○を選んで、
例えば135番の波動関数を描く場合は、W135-3D.scaを選択し、
【開く(O)】をクリックします。
※電子密度を描く場合はCHG3D.scaを、静電ポテンシャルを描く場合はPOT3D.scaを選択します。
- ※補足
VEND: Menu の【Preferences】をクリックし、
Preferencesウィンドウの【Other settings】の【Default ext.:】を
“scat”にして【Save as default】ボタンを押すと、
VENUS\Programs\preferences_VEND.iniファイルが更新され、
次回からはVEND: Menu の 【Open】をクリックしたとき、
Format: のプルダウンメニューで、【SCAT (*.scat, *.sca)】が
初期状態で表示されるようになります。
- ◎13.Dialogウィンドウの【Properties + 】をクリックし、
【Isosurfaces】をクリックし、Isosurface level: の値を、
波動関数の場合は例えば0.01a0-3/2や0.005a0-3/2など、小さい値にして、
【Apply】をクリックします。
例えば、初期値に、Isosurface level: 0.2488a0-3/2となって
いる場合は、Isosurface level: 0.02a0-3/2と入力して、
【Apply】をクリックしてみてください。
※電子密度を描く場合は例えば0.02a0-3など、
静電ポテンシャルを描く場合は例えば0.5 Ryなど、
それぞれ適当な数値を入れて【Apply】をクリックします。
※通常、波動関数ファイルには正の値と負の値が、
電子密度ファイルには正の値のみが、
静電ポテンシャルファイルには正の値と負の値が含まれています。
(陽イオンの計算の場合などは、静電ポテンシャルファイルにほとんど負の値しか含まれていない場合もあります。)
この値を小さくすればするほど、波動関数(または電子密度、静電ポテンシャル)の拡がりは大きく表示されます。
この値を大きくすればするほど、波動関数(または電子密度、静電ポテンシャル)の拡がりは小さく表示されます。
さらに、Smoothingの【More】をクリックすれば、
波動関数(または電子密度、静電ポテンシャル)の等値表面がスムージングされて、滑らかになります。
さらに、Opacity (%): 100
となっている箇所が右下の欄にありますので、これを例えば、
Opacity (%): 50
として、【Apply】をクリックします。
そうしますと、50 %ぐらい透明に透けて見えるようになります。
Opacityは不透明度という意味です。
100 %が普通の状態で、内部は全く見えませんが、
30 %だと内部がかなり透けて見えるようになります。
とりあえず、50 %ぐらいでいいと思います。
【OK】をクリックしてIsosurfacesウィンドウを閉じます。
- ◎14.VEND: Menu の【VICS】をクリックし、
ファイルの場所(I): にc:\dvxa\calc\○○を選んで、
VICSで保存したVICSファイル(例えばcomplex.vcs)を選択、
【開く(O)】をクリックします。
そうしますと、Dialogウィンドウに
□Show model
という行が追加されますので、□Show modelの□をクリックして“レ”を付けてやります。
この状態で、ボール&スティック表示の分子モデルが表示されているかと思います。
さらに、Dialogウィンドウの【Model】をクリックし、
例えばModelの欄の
◎Ball-and-stick
○Stick
を、
○Ball-and-stick
◎Stick
にしてやって【OK】をクリックすれば、分子モデルはスティック表示になります。
- ◎15.c04dで指定した描画範囲(例えば20a0×20a0×20a0)がUnit cellとして白い白線で表示されているかと思いますが、
もしこの白い白線が不要でしたら、Dialogウィンドウの【Unit cell】をクリックし、□Show unit cell edgesの選択をはずしてやれば、消すことができます。
- ◎16.Graphicsウィンドウをマウスの左ドラッグで好きな位置に回転させてやり、
VEND: Menuの【Export】をクリックし、
Export Imageウィンドウで適当な名前をつけて
ファイルの種類(T):
BMP (*.bmp)
EPS (*.eps)
JPEG (*.jpg)
JPEG 2000 (*.jp2)
PPM (*.ppm)
RAW (*.raw)
RGB (*.rgb)
TGA (*.tga)
TIFF (*.tif)
EPS (vector data) (*.eps)
の中から、適当なファイル書式を選択して保存してやれば、
各種お絵かきソフト、ワープロ、プレゼンテーションソフト
等に持っていけるかと思います。
以下に、以上の操作で描いた、ある金の錯体の分子軌道を以下に示します(JPEGファイルです)。
CIFからこの図を得るまで、DV-Xα計算も含めてなんとたったの1時間程度の作業でした(サンプル点数は少なめにした計算ですが)。

【電子密度の図】
※図をクリックすると大きな画像をご覧いただけます【101 KB】

【113番目の分子軌道の図】
※図をクリックすると大きな画像をご覧いただけます【167 KB】

【114番目の分子軌道の図】
※図をクリックすると大きな画像をご覧いただけます【187 KB】
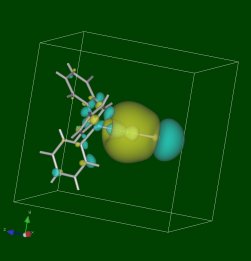
【115番目の分子軌道の図】
※図をクリックすると大きな画像をご覧いただけます【163 KB】

【116番目の分子軌道の図】
※図をクリックすると大きな画像をご覧いただけます【157 KB】

【117番目の分子軌道の図】
※図をクリックすると大きな画像をご覧いただけます【173 KB】

【118番目の分子軌道の図】
※図をクリックすると大きな画像をご覧いただけます【193 KB】

【119番目の分子軌道の図】
※図をクリックすると大きな画像をご覧いただけます【187 KB】
※その他いろいろな錯体の計算(DV-Xα+VENUS)結果は、こちらをご覧ください。
→ もくじに戻る
坂根ページへ/化学科ページへ
岡山理科大学 理学部 化学科 坂根弦太
ご意見・ご質問は下記まで
gsakane@chem.ous.ac.jp







